These are KEI’s February 17, 2013 comments on the the Bayer appeal of the compulsory license on Nexavar patents.
Comments on the the Bayer appeal of the compulsory license on Nexavar patents.
February 17, 2013
The dispute is Bayer Corporation v Natco Pharma Limited, and is being heardbefore the Intellectual Property Appellate Board At Chennai (O.R.A. no. 35/PT/2012).
1. Knowledge Ecology International (KEI) is a non-profit, non-governmental organization that searches for better outcomes, including new solutions to the management of knowledge resources. In particular, KEI is focused on the management of these resources in the context of social justice and human rights. KEI is drawn to areas where current businesses models and practices by businesses, governments or other actors fail to adequately address social needs or where there are opportunities for substantial improvements. Among other areas, KEI has expertise in access to medical technologies, including extensive experience in analyzing the costs of drug development and the role of generic competition in driving down prices so that consumers can purchase affordable medicines. Additionally, KEI has expertise in the Agreement on Trade Related Aspects of Intellectual Property Rights (TRIPS), including the use of the flexibilities retained by members to this agreement.
2. This statement covers three topics in the current case. First, it reviews the relationship between the World Trade Organization (WTO) rules on intellectual property rights, including in particular the WTO Agreement on Trade Related Aspects of Intellectual Property Rights, known as the TRIPS Agreement, and the decision by the Indian Controller of Patents to issue a compulsory license. Second, the statement discusses the evidence regarding the cost of research and development for the development of Nexavar/sorafenib, and in refutes several of the aggressive, and sometimes contradictory, claims made by Bayer in the proceedings, and discusses the relevance of research and development costs in making a product available at a reasonably affordable price. Finally, the statement discusses the decision by the Controller of Patents to set the appropriate royalty at six percent of the generic price.
Compulsory Licensing
WTO Rules Regarding Compulsory Licensing of Patents on Drugs and Other Medical Inventions
3. Under the World Trade Organization (WTO) Agreement on Trade-Related Aspects of Intellectual Property Rights (TRIPS), which came into force on January 1, 1995, members agree to certain minimum standards for intellectual property. While the agreement sets minimum standards of protection, members also retained a number of flexibilities known as “TRIPS flexibilities.” The TRIPS Agreement set forth specific provisions that recognize and in some cases regulate the granting of compulsory licenses and other non-voluntary uses of patents.
4. While the TRIPS Agreement provides a set of general rules, it also leaves much of the implementation details to members. The Article 1.1 of the TRIPS Agreement states that, “Members shall be free to determine the appropriate method of implementing the provisions of this Agreement within their own legal system and practice.”
5. Several provisions of the TRIPS Agreement are relevant to the issue of compulsory licensing. Article 31 of the TRIPS Agreement is one of the several provisions that provides for the possibility of limitations on exclusive rights. In Article 31, governments may provide for use without the authorization of the right holder, subject to certain rules. In Article 31(b), a member may issue a compulsory license, but only after an attempt has been made to obtain a license under reasonable terms and conditions within a reasonable period of time. However, this general requirement is subject to several exceptions, including the waiver in Article 31(b) for three separate cases: a national emergency, other circumstances of extreme urgency, or public non-commercial use. An additional waiver exists under Article 31(k), to “remedy a practice determined after judicial or administrative process to be anti-competitive.” Additional possibilities for granting compulsory licenses exist under Article 30, as well as Article 44, when injunctions are not granted to stop infringements. Each of these Articles sets its own rules, but taken together, they provide considerable room for governments to grant compulsory licenses, particularly when the compulsory license involves the payment of a royalty to the patent holder.
6. A recent trilateral report issued by the World Trade Organization (WTO), the World Intellectual Property Organization (WIPO), and the World Health Organization (WHO) on “Promoting Access to Medical Technologies and Innovation” confirmed the flexibilities members retain under TRIPS to issue compulsory licenses and the circumstances under which a government may grant a license. The report notes as a key point that “WTO members are free to determine the grounds for granting compulsory licences. Such grounds can include public interest in general and are not limited to public health emergencies.” WTO, WIPO and WHO, Promoting Access to Medical Technologies and Innovation (2013) at 171.
7. In 2001, members of the WTO negotiated what is known as the Doha Declaration on the TRIPS Agreement and Public Health (hereinafter “Doha Declaration”). The decision was adopted during a WTO ministerial meeting, on November 14, 2001. The Declaration did not create new rights for members, but instead clarified and confirmed the existing flexibilities under the TRIPS Agreement.
8. In particular, Paragraph 4 of the Doha Declaration reads:
We agree that the TRIPS Agreement does not and should not prevent members from taking measures to protect public health. Accordingly, while reiterating our commitment to the TRIPS Agreement, we affirm that the Agreement can and should be interpreted and implemented in a manner supportive of WTO members’ right to protect public health and, in particular, to promote access to medicines for all.
In this connection, we reaffirm the right of WTO members to use, to the full, the provisions in the TRIPS Agreement, which provide flexibility for this purpose (emphasis added).
It should first be noted that the Doha Declaration confirms that members have the right “to use, to the full” the flexibilities of the TRIPS Agreement, and that the “Agreement can and should be interpreted . . . in a manner supportive of WTO member’s right to protect health and, in particular, to promote access to medicines for all.” Secondly, it is important to recognize that the Doha Declaration puts an obligation on WTO members that TRIPS “should be . . . implemented” in a manner that promotes access to medicine for all.
9. The rights and obligations of members “to promote access to medicines for all” has been reaffirmed not only in the Doha Declaration, but also in other international documents including, for example, the World Health Organization’s Global strategy and plan of action on public health, innovation and intellectual property. 61st World Health Assembly, WHA Resolution 61.21 (24 May 2008).
10. The Doha Declaration further clarified that one of the important flexibilities under the TRIPS Agreement is “Each members has the right to grant compulsory licences and the freedom to determine the grounds upon which such licences are granted.” Doha Declaration at ¶5(b) (emphasis added). Granting of a compulsory license is well within the rights preserved by members in full accordance with the rights and obligations under the TRIPS Agreement. As the Doha Declaration affirms and clarifies, governments have an obligation to implement the TRIPS Agreement, including the use of TRIPS flexibilities, to protect public health and promote “access to medicine for all.”
11. The WTO, WIPO and WHO report notes that the freedom to determine the grounds upon which compulsory licenses are granted are “not limited to emergencies or other urgent situations, as is sometimes mistakenly believed” and that a “range of grounds” exist in national laws to issue compulsory licenses including:
Non-working or insufficient working: Many countries provide that where a patentee fails to work a patent in its jurisdiction, or where such working by the patentee is insufficient, a compulsory licence may be granted, provided that all other requirements are met. Some national laws simply state that if a patentee is not working the invention, or is not sufficiently working the invention without any legitimate justification, a third party may request a compulsory licence. In some countries, the laws provide detailed provisions clarifying the circumstances that may be applicable. Such clarifications include the types of activities by the patentee that are considered as “working”, in particular, whether importation of the patented invention is considered as “working” in the country or not, and the situations under which working by the patentee is not considered “sufficient.”
Anti-competitive practices: Some countries provide specific provisions under the patent law that allow the granting of a compulsory licence, in order to remedy an anti-competitive practice engaged in by the patentee. In certain countries, such as the United States, such a remedy is regulated in the competition (antitrust) law, under which compulsory licences may be granted by a competition authority (e.g. the US Federal Trade Commission) where it finds that it is an appropriate remedial action against an adjudicated anti-competitive practice.
Public interest: Many countries allow the granting of compulsory licences on grounds of public interest, without further defining the term. Others mention specific grounds, in particular, national emergencies and circumstances of extreme urgency, national security and public health in general. However, a national emergency or extreme urgency is not a prerequisite requirement for a compulsory licence under the TRIPS Agreement. Public interest could also include the non-availability of the patented product, such that reasonable needs of the public are not being met. In some cases, the laws refer to more specific health-related situations, such as a compulsory licence on a patent relating to diagnostics, or on a patent concerning a biotechnological research tool. Health-specific grounds can, for example, be found in France and Morocco. Under provisions on the licence d’office dans l’intérêt de la santé publique, the health minister can seek the grant of a compulsory licence if the product or method is made available by the right holder in insufficient quantity or unsatisfactory quality, or if the prices charged are abnormally high.
Dependent and blocking patents: Many countries provide for the possibility of requesting a compulsory licence where a patent (second or “dependent” patent) cannot be exploited without infringing another patent (first or “blocking” patent). Article 31(l) of the TRIPS Agreement provides that such compulsory licences can only be granted if the second invention is an important technical advance of considerable economic significance and that, where a compulsory licence is granted to the holder of a second (dependent) patent to use a first (blocking) patent, the holder of the first patent shall also have a right to a cross-licence to use the second patent.
Government use: A number of national laws explicitly entitles the government, or a third party authorized by the government, to use a patented invention without authorization of the patent holder. The grounds may vary but typically relate to public policy objectives such as national security or health. A specific authorization may be needed to use a patented technology, or the legal system may limit the scope of remedies that are available when a patent is infringed in the performance of a task authorized by the government. WTO, WIPO and WHO, “Promoting Access to Medical Technologies and Innovation” (2013).
Remedies to Patent Abuses in the TRIPS Agreement.
12. Particularly relevant to the dispute in the present case are the provisions in the TRIPS Agreement that concern the control of and remedy to abuses of rights, including anti-competitive practices. The case involves an allegation of abuses with regard to the pricing of the product, but also the failure to manufacture the product in India.
13. Compulsory licenses to remedy anti-competitive practices such as excessive prices are given special attention and broad latitude in the TRIPS Agreement, as are provisions dealing with working and the transfer of technology. Specific provisions in the TRIPS Agreement on these topics include Article 2 (which refers to Articles 1-12,19 of the Paris Convention), Article 8.2, Article 31(k), and Article 40.
14. Article 5A(2) of the Paris Convention provides that, “Each country of the Union shall have the right to take legislative measures providing for the grant of compulsory licences to prevent the abuses which might result from the exercise of the exclusive rights conferred by the patent, for example, failure to work.”
15. In Part II, Article 8.2 of TRIPS provides that appropriate measures, consistent with the Agreement, “may be needed to prevent the abuse of intellectual property rights by right holders or the resort to practices which unreasonably restrain trade or adversely affect the international transfer of technology.”
16. Article 31(k) of TRIPS deals the cases seeking to “remedy a practice determined after judicial or administrative process to be anticompetitive.” As noted above, Article 31(k) specifically provides a waiver of the general requirement for a prior effort to obtain a voluntary license on reasonable commercial terms, and also waives restrictions on the exports of products. Article 31(k) further states that, “the need to correct anti-competitive practices may be taken into account in determining the amount of remuneration in such cases.”
17. Article 40 of TRIPS is located in Part II, Section 8, under the heading: “Control of Anti-Competitive Practices in Contractual Licences.” Article 40.1 provides that “Members agree that some licensing practices or conditions pertaining to intellectual property rights which restrain competition may have adverse effects on trade and may impede the transfer and dissemination of technology.” Article 40.2 specifies that “Nothing in this Agreement shall prevent Members from specifying in their legislation licensing practices or conditions that may in particular cases constitute an abuse of intellectual property rights having an adverse effect on competition in the relevant market.”
Limits on Remedies from Infringement
18. One of the issues in the current case involves the alleged infringement of Bayer’s patents by CIPLA, following failures of Bayer to obtain an injunction against CIPLA. We note that Article 44 of the TRIPS Agreement provides for judicial discretion in granting injunctions to prevent such infringement under Article 44.1, the provision relating to the facts in the Bayer/CIPLA dispute. It also permits for broader limits on injunctions under Article 44.2, which provides for the possibility of eliminating the possibility of obtaining an injunction when certain conditions are met with regard to compensation or remuneration. Notably, in the United States, there is a rapidly expanding use of such flexibilities in decisions where injunctive relief is denied by a judge under the new standards for injunctions set out by in eBay v MercExchange, 547 U.S. 388 (2006), or in various United States statutes including 28 U.S.C. §1498 in the customs statute and 35 U.S.C. §271(3)(2) which places restrictions on injunctions in cases involving early working of patents or failures to provide competitors with relevant information on patents for biologic drugs.
19. In the case of the aforementioned statutes, the United States takes advantage of the ability to grant compulsory license rather than enforce injunctions for infringement, under the policy space provided by Article 44 of the TRIPS Agreement. Article 44.2 provides that
Notwithstanding the other provisions of this Part and provided that the provisions of Part II specifically addressing use by governments, or by third parties authorized by a government, without the authorization of the right holder are complied with, Members may limit the remedies available against such use to payment of remuneration in accordance with subparagraph (h) of Article 31. In other cases, the remedies under this Part shall apply or, where these remedies are inconsistent with a Member’s law, declaratory judgments and adequate compensation shall be available.
20. Where injunctions are not enforced and the remedy is limited to payment of remuneration, the effect is a compulsory license under Article 44 rather than Article 31 of the TRIPS Agreement. This type of compulsory license, also known as a “judicial compulsory license,” is common practice in the United States and has been granted for a wide range of patented products, including, for example, for medical devices.
21. On January 8, 2013, the U.S. Patent and Trademark Office and the U.S. Department of Justice issued a joint “Policy Statement on Remedies for Standards-Essential Patents Subject to Voluntary F/RAND Commitments.” This statement was designed to greatly expand the use of compulsory licenses under Article 44 of the TRIPS, by withholding injunctions for infringements in cases involving “standards-essential patents.” The policy was designed to protect “the public health and welfare, competitive conditions in the United States economy, the production of like or directly competitive articles in the United States, and United States consumers,” and cited 19 U.S.C. § 1337(d)(1). The statement notes the harms caused by the exclusive patent rights, including the high costs of switching to new products, higher prices for products, and “unwarranted higher royalties . . . passed on to consumers in the form of higher prices.” The statement noted that when administering the U.S. law:
public interest factors “are not meant to be given mere lip service,” but rather “public health and welfare and the assurance of competitive conditions in the United States economy must be the overriding considerations in the administration of this statute.”
Lack of domestic manufacturing as a grounds for compulsory license
22. Bayer cites Article 27.1 of the TRIPS Agreement’s which provides that “patents shall be available and patent rights enjoyable without discrimination as to the place of invention, the field of technology and whether products are imported or locally produced” in its criticism of the controller’s finding that the lack of working of the patent included a failure to manufacture the product in India. The WTO rules on this issue are not obvious, given the conflicting provisions in the TRIPS Agreement with regard to the importance of the transfer of technology in TRIPS Agreement’s Articles 8 and 40, and the references to Article 5 of the Paris Convention which also deal with importation and working of the patent. In the one WTO case where this issue was raised by the United States in the context of a statutory ground in Brazil, the case was withdrawn and settled without an agreement by Brazil to amend its statute.[1] The fact that no WTO dispute has been litigated to resolution on this issue reflects the weakness of the legal case of those who claim local working obligations violate the TRIPS. Moreover, several countries including the United States have policies which favour domestic manufacturing of patented goods in some cases, and there is recognition in several jurisdictions that local manufacturing can have an impact on domestic employment, technology transfer or the supply of an essential good. For example, in the United States, a patent injunction was not made available to stop the infringement of a patent on a medical device when the injunction would have caused a shift of the manufacturing from the United States to Mexico.[2] Additionally, several countries, including the United States, have indicated that the local manufacturing of some medicines will enhance national access in periods of a global health emergency.[3]
23. In its 2004 Handbook on Intellectual Property, the UN’s World Intellectual Property Organization (WIPO) provides an extensive discussion of the Paris Convention provisions on compulsory licenses for failure to work a patent, including the following passages:
5.45 Compulsory licenses on the ground of failure to work or insufficient working are the most common kind of coercive measure against the patent owner to prevent abuses of the rights conferred by the patent for invention. They are expressly dealt with by Article 5A.
5.46 The main argument for enforcing working of the invention in a particular country is the consideration that, in order to promote the industrialization of the country, patents for invention should not be used merely to block the working of the invention in the country or to monopolize importation of the patented article by the patent owner. They should rather be used to introduce the use of the new technology into the country. Whether the patent owner can really be expected to do so, is first of all an economic consideration and then also a question of time. Working in all countries is generally not economical. Moreover, it is generally recognized that immediate working in all countries is impossible. Article 5A therefore tries to strike a balance between these conflicting interests. [WIPO Intellectual Property Handbook, Wipo Publication No. 489 (E), ISBN 978-92-805-1291-5, WIPO 2004, Second Edition, Reprinted 2008]
24. An example of a specific requirement for domestic manufacturing is the United States’ Bayh-Dole Act, which applies to patented inventions that benefit from U.S. government research and development funding. This requirement includes the obligation found in 35 U.S.C. §204, “Preference for United States industry” which states:
Notwithstanding any other provision of this chapter, no small business firm or non-profit organization which receives title to any subject invention and no assignee of any such small business firm or non-profit organization shall grant to any person the exclusive right to use or sell any subject invention in the United States unless such person agrees that any products embodying the subject invention or produced through the use of the subject invention will be manufactured substantially in the United States. However, in individual cases, the requirement for such an agreement may be waived by the Federal agency under whose funding agreement the invention was made upon a showing by the small business firm, non-profit organization, or assignee that reasonable but unsuccessful efforts have been made to grant licenses on similar terms to potential licensees that would be likely to manufacture substantially in the United States or that under the circumstances domestic manufacture is not commercially feasible.
A failure to manufacture the patented invention is a grounds for a compulsory license under 35 U.S.C. §203 provision for march-in rights.
25. Another example of a domestic working obligation is found in the United States law on “Energy Storage Competitiveness.” Under 42 U.S.C. §17231(h)(7), the government has powers to require the license of patents in order to “advance the capability of the United States to successfully compete in global energy storage markets.”
26. Over the past three years, there has been a global shortage of drugs to treat Fabry’s disease. In the United States, Genzyme was the sole supplier of a drug to treat this disease, and the company experienced a series of manufacturing failures that led to life threatening and health impairing rationing of medicines to U.S. patients. In Germany, a compulsory licensing case involving the Europe based Shire Pharmaceuticals and the U.S. patent holder led to an agreement that permitted two suppliers for the European market. As a consequence of the German compulsory licensing case and the competition between suppliers, patients suffering from Fabry’s disease in Europe received full doses of medicines.
27. Apart from the merits of local manufacturing in promoting national access to medicines and the transfer of technology, Bayer does not have the standing to pursue litigation before the WTO on such matters — only WTO member states may seek a remedy to a violation of the TRIPS.
The Indian Statute
28. Section 84 of the India Patent Act governs compulsory licenses and reads
(1) At any time after the expiration of three years from the date of the grant of a patent, any person interested may make an application to the Controller for grant of compulsory license on patent on any of the following grounds, namely—
- (a) that the reasonable requirements of the public with respect to the patented invention have not been satisfied, or
- (b) that the patented invention is not available to the public at a reasonably affordable price, or
- (c) that the patented invention is not worked in the territory of India.
The Indian statute is well within the contours of the policy space provided by the WTO TRIPS Agreement. Note that Paragraphs 6-20 of James Packard Love’s affidavit in the present case discuss the standards for “reasonably affordable price.” James Packard Love, Affidavit, Natco Pharma Ltd. v. Bayer Corporation, C.L. No. 1/2011 (13 February 2013).
Costs of Research and Development
29. Patent holders frequently assert the high costs of drug development as a justification for high prices and advocacy for higher patent protection. Unfortunately, the actual costs of drug development are often not disclosed to the public, and company or third party estimates can be speculative, controversial and self-serving.
30. A 2011 peer reviewed survey of estimates of drug research and development costs published in the journal Health Affairs notes,
Despite three decades of research in this area, no published estimate of the cost of developing a new drug can be considered a gold standard. Existing studies vary in their methods, data sources, samples, and therefore estimates. While some methods are methodologically strong and some findings have been widely cited, the fact that the data and even the subjects of investigation are kept secret make it impossible to assess validity and reliability. Steve Morgan, Paul Grootendorst, Joel Lexchin, Colleen Cunningham, and Devon Greyson, The Costs of Drug Development: A systematic review, 100 Health Pol’y 1 (2011).
31. In oral arguments in the present case, Bayer asserted it has spent “2 billion Euros” to develop Nexavar/sorafenib. To make this argument, Bayer presented a January 9, 2013 affidavit from Harold Dinter, which made the claim that from 1999 to 2005 Bayer had spent “2 billion euros (approximately US$ 2.5 billion) in the identification and development of anti-cancer molecules leading to the successful approval of Nexavar/sorafenib in 2005.” Dinter did not provide detailed support for the numbers, but stated they were based upon Bayer’s general research and development outlays for anti-cancer drugs, including but not limited to Nexavar/sorafenib, and that the estimate was supported by a new December 2012 study by Jorge Mestre-Ferrandiz, Jon Sussex and Adrian Towse, published by the Office of Health Economics (OHE), as well as earlier studies co-authored by Joseph DiMasi.
32. Bayer had declined to present its actual outlays that were specific to Nexavar/sorafenib development, even though Onyx Pharmaceuticals, Bayer’s partner in the development of Nexavar, has made extensive disclosures of R&D costs to the US Securities and Exchange Commission (SEC) in its annual 10-K reports.
33. The $2.5 billion figure Bayer presented was more than a billion higher than the December 2012 Mestre-Ferrandiz, Jon Sussex and Adrian Towse estimate of the average cost of developing a new drug, and about $1.7 billion higher than DiMasi’s widely cited 2003 paper, which put the risk and capital cost adjusted cost of the development of a new drug at $802 million. There are several reasons why the court should disregard Bayer’s extremely aggressive assertions of R&D costs.
34. First, there numerous inconsistencies between both the OHE and DiMasi estimates and the facts in the Nexavar/sorafenib case. When read and put into perspective, the OHE and DiMasi papers do not support Bayer’s claims of 2 billion Euros in R&D outlays, but instead undermine those claims. A discussion of those studies and their relevance to the Nexavar/sorafenib case follows.
35. Among the studies relied upon by Bayer in claiming the 2 billion Euro outlays are several authored by or based upon the work of Joseph DiMasi, a researcher from Tufts University who works extensively as a consultant to the pharmaceutical industry. The most important DiMasi paper, which has been used as the basis of many subsequent estimates by DiMasi and others, was a 2003 paper published the Journal of Health Economics. J.A. DiMasi et al., 22 Journal of Health Economics 151–185 (2003). This study, first publicized by the CEO of Merck, before publication in a journal,[4] was based on data on the costs of clinical testing of drugs from a confidential survey of big pharmaceutical companies. That survey excluded data from the development of Nexavar/sorafenib, a drug that was first registered by the FDA in 2005.
36. In the 2003 DiMasi paper, DiMasi described the drugs development projects in his sample as:
Ten multinational pharmaceutical firms, including both foreign and US-owned firms, provided data through a confidential survey of their new drug R&D costs. Data were collected on clinical phase costs for a randomly selected sample of the investigational drugs of the firms participating in the survey. The sample was taken from a Tufts Center for the Study of Drug Development (CSDD) database of investigational compounds. Cost and time data were also collected for expenditures on the kind of animal testing that often occurs concurrently with clinical trials. The compounds chosen were all self-originated; that is, their development up to initial regulatory marketing approval was conducted under the auspices of the surveyed firm. Licensed-in compounds were excluded because non-survey firms would have conducted portions of the R&D (footnotes omitted).
While there are few details available of the drugs used in the survey, DiMasi has indicated that the sample did not focus on Orphan Drugs, or drugs from small biotech firms.
37. Bayer also relied upon a 2012 study by Jorge Mestre-Ferrandiz, Jon Sussex and Adrian Towse that was published by the Office of Health Economics (OHE). Despite its name, the OHE is not a government body, but rather, an industry-funded private consulting firm and AstraZeneca paid for the study.
38. Several factors contribute to the cost of drug development including the origin of the medicine, the time involved in development, the size of the clinical studies, and the subsidies by various governments in the drug development process. In all of these factors the known facts in the Nexavar/sorafenib case do not support the aggressive Bayer assertions regarding the cost of drug development. Moreover, there are objective data points on research and development costs from Bayer’s partner in Nexavar/sorafenib’s development that do not support Bayer’s claims.
39. DiMasi’s 2003 estimate of $802 million for the cost of developing a new drug was built upon much smaller estimates of the cost of “out of pocket” outlays on particular drugs. In particular, most of the data was extrapolated from outlays on Phase I, II and III clinical testing. DiMasi did not have firm outlays on pre-clinical testing costs, other than animal testing, and he adjusted the out-of-pocket costs for his estimates of the risks in the development process as well as the lost profits investors would forego during the development process.
40. The 2003 DiMasi estimates found that the average costs of Phase I, II and III tests for the sample were $125 million, and the median costs were $92.9 million, in 2000 USD. See Table 1 at p. 162.
| Phase | Mean | Median | Standard Deviation |
| Phase I | $15.2 | $13.9 | $12.8 |
| Phase II | $23.5 | $17.0 | $22.1 |
| Phase III | $86.3 | $62.0 | $60.6 |
| Sum of Phases I-III | $125.0 | $92.9 | |
The fact that the median is lower than the mean, and standard deviation for costs is roughly the size of the median cost estimates, reminds that drug development costs are highly variable, even within DiMasi selective sample from 10 firms, 8 of which were among the largest 20 drug companies. In footnote 41, page 177, the average number of patients in clinical trials is reported to be 5,303. The average cost per patient of $23,572 included not only direct costs, but also allocations of company overheads and other indirect costs.
41. The 2012 OHE estimates are also based upon confidential surveys, and assume higher estimates of clinical testing, in part because estimates are expressed in 2011 dollars, but also because of different assumptions in other areas. The OHE report estimated that within their sample, the out-of-pocket costs on development ranged from a high of $563.8 million and a low of $21.81 million, for an average of $234.7 million and a median of $151.5 million.[5] With the high cost product more than 26 times as expensive as the low cost product, one can appreciate how difficult it is to draw conclusions from selective samples or even means in general. What then can be drawn from these industry consultant estimates when considering the known facts from the Nexavar/sorafenib case?
Nexavar/sorafenib is a Licensed-In Product
42. Nexavar/sorafenib is a licensed-in drug, developed in a partnership with Onyx Pharmaceuticals, a small pharmaceutical company.
43. Page 156 of DiMasi’s 2003 paper notes, “Licensed-in compounds were excluded because non-survey firms would have conducted portions of the R&D.” DiMasi further notes on page 179 that:
while PhRMA has traditionally disaggregated its reported R&D expenditure data into expenditures on new drugs and expenditures on improvements to existing drugs, it has not gathered information on how expenditures on new drugs can be further decomposed into expenditures on self-originated and on licensed-in new drugs. Our R&D cost estimates are for self-originated drugs, and a substantial portion of the R&D expenditures on licensed-in drugs are likely missing from the PhRMA data.
44. In their discussion of “Self-Originated versus Licensed-In” compounds, the OHE authors note that DiMasi’s subsequent research on licensed-in compounds suggests that risks are lower for licensed-in products, and that DiMasi’s 2003 fully-loaded-risk-adjusted estimate of $802 million for self-originated compounds would fall to $640 million in 2003, based upon on the higher success rate alone, when the analysis was applied to licensed-in compounds.
DiMasi et al (2010) . . . estimate a higher overall clinical approval success rate for licensed-in versus self-originated drugs: 27% versus 16%. However, the two groups of medicines had identical success rates for both Phase III (64%) and regulatory review (93%). Higher overall clinical approval success rate implies, other things constant, lower total R&D costs per approved drug; for instance, and using the results from DiMasi et al (2003), the R&D cost per approved drug with the higher 27% success rate would fall to US$640m (2003 prices), assuming other things stay constant, as compared with the original estimate of US$802m . . . OHE at 53.
45. Note that for both the DiMasi and OHE methodologies, the bigger numbers reflected the overall costs and are “fully loaded” with regard to overhead costs, pre-clinical research, all risks of failures, and an aggressive estimate of the cost of capital. Thus, the OHE report states that when a single adjustment is made to take into account the fact that Nexavar/sorafenib is a licensed-in compound, the OHE would put the fully loaded and risk adjusted costs at $640 million, in 2003 prices.[6] This figure reflects just 25.6 percent of the Bayer claim of $2.5 billion. This result calls into question why Bayer chose to put this document into the record, because it clearly rebuts its own claims. Furthermore, additional adjustments for other factors must be made to the $640 million figure, thus shrinking the costs even more.
Nexavar/sorafenib’s development time was quicker
46. In his 2003 paper, DiMasi estimates the average time between the beginning of clinical testing and FDA approval is 90.3 months. Nexavar/sorafenib began clinical testing in 2000 and was approved by the FDA December 2005, a significantly shorter period than the average used in the DiMasi estimate. The difference in the period of time for testing, and the fact that most of the spending on Nexavar/sorafenib took place in 2004 and 2005, is important because DiMasi and the OHE both add an 11 percent (or more) after inflation compound real cost of capital to adjust the out-of-pocket costs upwards.
Nexavar/sorafenib benefited from Orphan Drug Status
47. DiMasi avoided orphan products in his 2003 paper, and stated, “For development as a whole, it is highly likely therefore that the share of R&D expenditures for which the orphan drug credit was applicable for traditional large multinational pharmaceutical firms is quite low.” DiMasi at 175. By avoiding orphan products, which have lower regulatory requirements, smaller trials, and a 50 percent tax credit available for qualifying clinical trials, DiMasi was able to provide higher estimates of drug development costs – one of the goals of the pharmaceutical firms that sponsor much of DiMasi’s work. Nexavar/sorafenib is not only a licensed-in product from a small company, but it has also received four orphan drug designations, including one for renal cell carcinoma.
48. When Nexavar/sorafenib received an orphan drug designation, the drug developer received the benefit of being able to directly reduce US federal income tax liability by 50 percent of the costs of the qualifying clinical trials, for that indication, via a special and very generous tax credit, and still deduct the remaining 50 percent of costs from federal income taxes. See IRS Form 8820, General Instructions. The Orphan Drug Tax Credit is equivalent to receiving a grant from the U.S. government, to cover half of the cost of the qualifying trials, not to mention other important benefits.[7] The orphan drug credit is particularly interesting in this case because Bayer received payments for 50 percent of its R&D costs on Nexavar/sorafenib from Onyx, and therefore, it is likely that Bayer incurred no cost of its own for the trials that qualified both for the 50 percent tax credit and the 50 percent funding from Onyx.
49. As an Orphan Drug, the size of the trials submitted to the FDA to establish the efficacy of the product were relatively small. For the 2005 approval, the two cite phase II trials involved 173 patients, and the Phase III trial cited involved between 769 and 900 patients, depending upon how it was reported. Even when adding Phase I trials, the numbers are far smaller than the 5303 average number of patients for the trials related to approval of a NME compound that were cited by DiMasi in 2003.
50. Using prevailing average costs for oncology trials at the time, Dr. Ruth Lopert has made an initial estimate that Bayer would not have spent more than $33.165 million for the trials that the FDA relied upon for its 2005 approval, before even taking into account the benefits of the 50 percent orphan drug tax credit. Lopert assumed costs of $18,833 per patient for Phase I trials, $19,643 per patient for Phase II trials, and $27,833 per patient for Phase III trials. For trials related to the 2005 approval of Nexavar/sorafenib, she estimated Bayer had 424 patients in Phase I trials, 173 patients in Phase II trials, and 769 patients in Phase III trials, for a total of 1366 patients in trials at an average cost of $24,279 per patient for all three phases. Note that Lopert’s estimate of $33.165 million for clinical testing costs was actually 52 percent higher than the lowest cost product in the 2012 OHE estimate.[8] If Bayer were to dispute Lopert’s estimate of clinical testing costs, it would have to challenge either the number of patients or the per-patient costs estimates.
51. Lopert’s estimate of $33.165 million was 26 percent of DiMasi’s higher estimate of $125 million for Phase I-III testing costs for non-orphan products — the difference largely explained by the lower number of patients in the Nexavar/sorafenib trials. Based upon her review of the FDA medical review and independent data on trials, Lopert found 1,366 patients in trials related to the 2005 FDA approval, which was just 26 percent of DiMasi’s 5303 average number of patients. The total Onyx/Bayer research and development outlays for the period up to the approval of Nexavar/sorafenib by the FDA in 2005 were higher because the total outlays included pre-clinical testing expenses and outlays on other uses of the drug that did not succeed, a 2000 patient expanded access trial that only benefited patients living in the United States, and the decision by Onyx to allocate costs for manufacturing facilities, overhead, extraordinary compensation to consultants and others as R&D costs.
Additional pre-approval Nexavar/sorafenib research and development projects.
52. Bayer and Onyx also undertook research on liver cancer, metastatic melanoma, or advanced skin cancer, non-small cell lung cancer, breast, prostate, ovarian and other cancers.
53. Apparently after receiving its 2004 Orphan Drug designation, Bayer and Onyx made Nexavar/sorafenib available to 2,000 patients in an expanded access program for kidney cancer, a factor that increased Bayer and Onyx’s R&D costs, but may also have qualified for the 50 percent orphan drug tax credit. Notably, Bayer has consistently refused to provide details of the orphan drug tax credit subsidies, even though the tax credit has certainly reduced its net costs.
54. In looking at research and development costs, the expanded access trials present a dilemma. While they involve research protocols because the drug is not yet approved for sale and cost money, it should be noted that not only do the trials have a much lower scientific value than the trials used to support a drug approval, but the expanded access program has no benefits to cancer patients outside of the United States, and it may easily be argued that they should therefore be excluded when considering costs in a proceeding in India.
Government Interest in Nexavar/sorafenib
55. In addition to receiving benefits such as the generous orphan drug tax credit, governments also took an interest in the product and, at least in the United States, the government funded trials on sorafenib. See James Packard Love, Affidavit at ¶¶33-34, Natco Pharma Ltd. v. Bayer Corporation, C.L. No. 1/2011 (13 February 2013).
Onyx’s 10-K reports of research and development outlays for Nexavar/sorafenib
56. As noted elsewhere, Bayer’s partner in the development of Nexavar/sorafenib is Onyx Pharmaceuticals. Onyx published annual estimates of its R&D spending on Nexavar/sorafenib. Beginning in 1994, Bayer entered into a drug development agreement with Onyx Pharmaceuticals. According to the Onyx 2002 10-K annual report filed with the U.S. Securities and Exchange Commission:
Effective February 1994, we established a research and development collaboration agreement with Bayer to discover, develop and market compounds that inhibit the function, or modulate the activity, of the Ras signalling pathway or that appropriately modulate the activity of this pathway to treat cancer and other diseases. Bayer and we concluded collaborative research under this agreement in 1999, and based on this research, a development candidate, BAY 43-9006, was identified.
57. From 1994 to 1999, Onyx reports that Bayer provided Onyx with $26.1 million for the Onyx work on the Ras signalling pathway. Beginning in 2000, Onyx and Bayer began another collaboration, aimed at the clinical testing and development of BAY 43-9006 and other products that had been identified in their joint research program. BAY 43-9006 was later given the generic name sorafenib, and the brand name Nexavar.
58. Onyx’s accounting for R&D outlays is quite inclusive, and the primary components beginning in 2000 were “clinical manufacturing costs, clinical trial expenses, consulting and other third-party costs, salaries and employee benefits, supplies and materials and allocations of various overhead and occupancy costs.”
59. After the FDA approved Nexavar in December 2005, Onyx provided an account of the cumulative research and development outlays on Nexavar/sorafenib from 2000 to the end of 2005. The Onyx 10K reports include not only the trials used to support the 2005 approval of Nexavar/sorafenib for the treatment of renal cell carcinoma, but also the outlays related to approval of Nexavar for any and all other uses, and the 2,000 patients in the expanded access program for kidney cancer.
60. According to the 2005 Onyx 10-K annual report: “Aggregate research and development costs-to-date through December 31, 2005 incurred by Onyx since fiscal year 2000 for the Nexavar project is $134.8 million.”
61. To summarize, note that the Bayer outlays on pre-clinical research related to Nexavar/sorafenib from 1994 to 1999 were reported by Onyx to be $26.1 million. The $26.1 million included research on several compounds in addition to the one now marketed as Nexavar/sorafenib. From 2000 onward, Bayer and Onyx split the R&D costs 50:50, and Onyx’s share of the R&D costs were $134.8 million. The outlays on the entire R&D program that lead to the 2005 approval of Nexavar/sorafenib for Kidney cancer were $26.1 + (134.8 x 2) = $295.7 million. Of the $295.7 million, only a fraction was spent on the development of Nexavar/sorafenib for kidney cancer, and some of that benefited from a 50 percent tax credit under the U.S. Orphan Drug Act.
62. To the put the entire $295.7 million into perspective, even ignoring cost reducing impact of the U.S. Orphan Drug tax credits, the R&D outlays reported by Onyx represent a little more than one quarter of the current global sales for Nexavar/sorafenib, bear in mind that it is a product that will maintain its monopoly in most markets through 2020. $295.7 is also just 11.8 percent of the $2.5 billion estimate that Bayer wants the IPAB to accept as its R&D costs.
63. In reviewing the data on drug development costs, it is difficult to understand how Bayer expects data on drug development costs to fit into the present case. Even if the $2.5 billion figure is accepted, the question of whether Bayer should be rewarded for being a high cost supplier of research and development — requiring more than one billion more than its peers, according to the OHE study – must be asked. Furthermore, will Bayer promise to give away Nexavar/sorafenib for free now that revenues have vastly exceeded even the most creative ways to blowing up the drug development cost figures? Further discussion of the revenues of Bayer for the product Nexavar/sorafenib is available in James Packard Love, Affidavit at ¶¶33-37, Natco Pharma Ltd. v. Bayer Corporation, C.L. No. 1/2011 (13 February 2013), a part of the record below.
Royalty-Setting
64. Bayer asks the court to overturn the decision to grant a royalty of 6 percent of the price of the generic product, and in its oral presentation of the appeal suggested a higher rate of 15 percent be applied to a price higher than the generic price.
65. Where a government issues a compulsory license, a patent holder has a right to receive adequate remuneration under Article 31(h) of the TRIPS Agreement, which provides, “the right holder shall be paid adequate remuneration in the circumstances of each case, taking into account the economic value of the authorisation.” Adequate remuneration is a relatively low standard, and the TRIPS Agreement even relaxes this standard in Article 31(k), which states, “The need to correct anti-competitive practices may be taken into account in determining the amount of remuneration.”
66. In a 2005 two UN agencies published a report on possible guidelines for setting royalties for non-voluntary authorizations to use patents. That report, authored by James Love, noted that “Countries retain broad authority . . . to set royalties according to systems of their choosing . . . Different countries may prefer different approaches to remuneration, based upon administrative capacity, resource constraints and policy objectives concerning access and innovation, among other factors.” World Health Organization (WHO) and United Nations Program on Development (UNDP), Remuneration Guidelines for Non-Voluntary Use of Patent on Medical Technologies, in Health Economics and Drugs, TCM Series No. 18.
67. The 2005 WHO/UNDP report identified three existing guidelines for setting royalties in cases involving compulsory licenses of patents, and proposed one additional method. These include the 1998 Japan Patent Office, the 2001 UNDP guidelines, the 2005 Canadian guidelines, and a new 2005 Tiered Royalty Method (TRM).
68. On 29 June 1998, the Japan Patent Office (JPO) reported new guidelines for determining royalty rates for licensing patents owned by the Japanese Government. While the guidelines were officially for setting royalties on government-owned patents, they were considered by some a de facto standard, and were influential in private sector compulsory licensing and infringement cases. The previous JPO guidelines were rates of 2 to 4 percent of net sales, and had not changed for 50 years. Under the revised guidelines, the royalties were 0 to 6 percent, with the exact rate determined by a sector of factors such as the expected profits, the utilization, exploration and increase/decrease factors. The initial base royalties were as follow:
| High | 4% (expected profits 30%) |
| Medium | 3% (expected profits 20%) |
| Low | 2% (expected profits 10%) |
As discussed in the WHO/UNDP report, these rates were then increased or decreased based upon other facts, but were never above 6 percent or below zero, and were applied to the price of the generic product.
69. In its 2001 Human Development Report, UNDP recommended that developing countries adopt royalty guidelines for compulsory licenses on pharmaceutical drugs, in order to provide greater transparency and predictability. UNDP specifically recommended that rates normally be set at 4 percent, and adjusted upwards as much as 2 percent for products of particular therapeutic value, or reduced as much as 2 percent when the development of the product had been partly supported with public funds, for a range of 2 to 6 percent, applied to the price of the generic product.
70. In 2005, Canada proposed royalty guidelines for the export of medicines under the Jean Chrétien Pledge to Africa Act, a law to implement the August 30, 2003 WTO decision to provide a waiver of Article 31(f) of the TRIPS Agreement. The 2005 Canadian royalty guidelines provided a sliding scale of 0 to 4 percent of the generic sales price. The exact rate depended upon the location of the importing market and the rank of the importing country in the UNDP Human Development Index. For India, the 2005 rate was set at just 1.2 percent.
71. The 2005 Tiered Royalty Method (TRM) was developed to provide a method of setting royalties independent of the price of the generic product. It required a reference market price for the drug where access was considered adequate and the price considered reasonable. From this reference market price, a base royalty was calculated from a base royalty rate (4 percent of the reference price in the 2005 WHO/UNDP report). Finally, this was adjusted to reflect the differences in the capacity to pay between countries. The use of the TRM is challenging for the Nexavar/soreafenib case for the following reason: The price of Nexavar/sorafenib is considered excessive and access limited even in high income country markets, so a direct reference price is not available. An indirect reference price could be constructed by using the World Bank standard of one unit of GDP per capita per DALY benefit. If one year of Nexavar/sorafenib treatment provided 1/4 year of extended life, and the base annual GDP per capita India was $1500, and the base royalty rate was 5 percent, the TRM royalty would be 1500 x .25 x .05 = $18.75 USD per year, or $1.62 per month.
72. By all four of the royalty guideline approaches, the Controller’s decision is on the high side as regards payments to Bayer.
73. Other data exists on royalty payments that suggest the 6 percent of generic prices figure is well within the range of reasonable outcomes.
74. In the April 6, 2007 book: Russell Parr, Royalty Rates for Licensing Intellectual Property, the median royalty rate for pharmaceutical and biotech licenses was reported at 5.1 percent.[9] Using Parr’s estimate of a 16.4 percent average industry profit rate for pharmaceutical and biotechnology companies, and the traditional 25 percent rule of thumb method[10] for estimating a reasonable royalty, the royalty rate would be 4.1 percent.
75. Another book by Russell Parr, Royalty Rates for Pharmaceuticals & Biotechnology, 7th Edition, presents a graph summarizing the royalty rate data found throughout the License Agreement section of his book. All royalty rates are presented as a percent of sales:
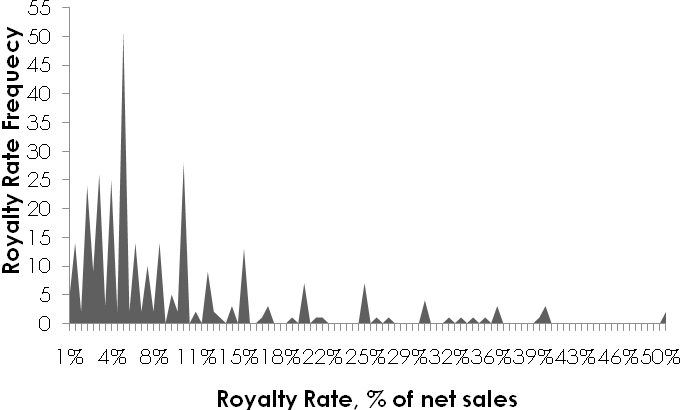
76. Note also that in the present case, the affidavit submitted by James Packard Love discusses in further detail the issues and factors of royalty-setting and was incorporated into the Controller’s decision below. James Packard Love, Affidavit at ¶¶42-57, Natco Pharma Ltd. v. Bayer Corporation, C.L. No. 1/2011 (13 February 2013).
77. Finally, when considering royalty rates, consider the purpose of the compulsory license. Bayer is being sanctioned for an abuse of the patent right, and in particular, for not making the patented product available at a reasonably affordable price. Assuming competition exists among generic suppliers, any royalty rate will likely result in higher prices to consumers. A very high royalty rate will itself defeat the purpose of the compulsory license and the India statute as regards expanding access to this drug for cancer.
Footnotes
1. DISPUTE SETTLEMENT: DISPUTE DS199, Brazil — Measures Affecting Patent Protection.
2. James Love, The CoreValve compulsory license on patent to treat aortic stenosis, September 1, 2011. /node/1218. Edwards Lifesciences AG and Edwards Lifesciences LLC, Plaintiffs, v. CoreValve, Inc. and, Medtronic CoreValve, LLC, Defendants. C.A. No. 08-91-GMS. United States District Court, D. Delaware., February 7, 2011. MEMORANDUM, Gregory M. Sleet, District Judge.
3. For example, see the video “Part 2, Leavitt and Rep Allen on patents and bird flu,” http://www.youtube.com/watch?v=q3VLE3MabcM, which reports an exchange during a November 8, 2005 hearing in the US Congress between Michael Leavitt, the Secretary of the U.S. Department of Health and Human Services, and Representative Tom Allen (D-Maine). The discussion focuses on the challenges of securing access to medicines for an influenza pandemic.
4. Gardiner Harris, Cost of Developing New Medicine Swelled To $802 Million, Research Study Reports, Wall Street Journal, December 3, 2001 http://online.wsj.com/article/SB1007336440403996240.html
5. OHE: Table 3-1, page 35.
6. Note that Nexavar was approved by the U.S. Food and Drug Administration in 2005.
7. See: Clark Herman, “The Orphan Drug Act at 30 Years: What’s Next?” PharmaExec.Com JANUARY 8, 2013. http://blog.pharmexec.com/2013/01/08/the-orphan-drug-act-at-30-years-what%E2%80%99s-next “The law granted seven-year patent exclusivity, tax credits equivalent to one half of the development cost (later modified to a fifteen-year carry-forward provision and a three-year carry-back that can be applied in any profitable year), direct grants, FDA fast-track approvals, and expanded access to patients under the Agency’s Investigational New Drug Program. The law was also later amended to waive FDA user fees established under PDUFA.”
8. OHE: Table 3-1, page 35.
9. Cited by William J. Murphy, John L. Orcutt and Paul C. Remus, Patent Valuation: Improving Decision Making through Analysis, Wiley Finance, May 8, 2012. Page 253.
10. Robert Goldscheider, The Classic 25% Rule And The Art Of Intellectual Property Licensing, Duke Law & Technology Review, No. 006, 2011. Also: See Murphy, Orcutt and Remus, Patent Valuation: Improving Decision Making through Analysis, Wiley Finance, May 8, 2012, which has a discussion of the 25 percent rule on page2 250 to 253.
Signed
James Packard Love
Krista L. Cox
Knowledge Ecology International
1621 Connecticut Ave. NW
Suite 500
Washington, DC 20002
United States of America
+1 202-332-2670