On August 20, 2018, KEI filed comments in response to the US Federal Trade Commission’s hearings and public comment process to address, “Competition and Consumer Protection in the 21st Century.” The goal of the process is to examine, “whether broad-based changes in the economy, evolving business practices, new technologies, or international developments might require adjustments to competition and consumer protection enforcement law, enforcement priorities, and policy.”
Respondents to the call for comments were asked to address the following topics:
- The state of antitrust and consumer protection law and enforcement, and their development, since the Pitofsky hearings;
- Competition and consumer protection issues in communication, information, and media technology networks;
- The identification and measurement of market power and entry barriers, and the evaluation of collusive, exclusionary, or predatory conduct or conduct that violates the consumer protection statutes enforced by the FTC, in markets featuring “platform” businesses;
- The intersection between privacy, big data, and competition;
- The Commission’s remedial authority to deter unfair and deceptive conduct in privacy and data security matters;
- Evaluating the competitive effects of corporate acquisitions and mergers;
- Evidence and analysis of monopsony power, including but not limited to, in labor markets;
- The role of intellectual property and competition policy in promoting innovation;
- The consumer welfare implications associated with the use of algorithmic decision tools, artificial intelligence, and predictive analytics;
- The interpretation and harmonization of state and federal statutes and regulations that prohibit unfair and deceptive acts and practices; and
- The agency’s investigation, enforcement, and remedial processes.
KEI submitted comments addressing topics 4, 6, 8, and 11. The text of KEI’s comments are below (with a few edits), and are also available as a PDF.
Regarding the hearing aspect of this process, the FTC will hold a two-day hearing September 13-14, 2018, the first of an expected 15-20 hearings to be held between September 2018 and January 2019. For more details and a schedule of the hearings, please visit: https://www.ftc.gov/policy/hearings-competition-consumer-protection
Competition and Consumer Protection in the 21st Century Hearings, Project Number P181201
Comments of Knowledge Ecology International
- The state of antitrust and consumer protection law and enforcement, and their development, since the Pitofsky hearings;
- Competition and consumer protection issues in communication, information and media technology networks;
- The identification and measurement of market power and entry barriers, and the evaluation of collusive, exclusionary, or predatory conduct or conduct that violates the consumer protection statutes enforced by the FTC, in markets featuring “platform” businesses;
- The intersection between privacy, big data, and competition;
Facebook and Twitter are two social networks that because of their reach and function are particularly important socially and politically. It is the view of KEI that both Facebook and Twitter have obtained monopolies by taking advantage of network effects and avoiding implementation of measures to permit rival clients to provide and receive status updates and media sharing with their users. If such interoperability were implemented, the services would have to compete on several dimensions including the user interfaces and policies on privacy.
Users of Facebook are now confronted with privacy policies they do not approve of, and Facebook and Twitter both face concerns about the regulation of content.
Breaking Facebook up into different services has been proposed, but typically by asking that Facebook divest certain non-core services it has acquired. The fact that alternatives to Facebook exist, including networks that Facebook itself owns and controls, provides some benefits to the public, but for some functions, the benefits of the service are related to its reach, so size is both a problem and a benefit.
Governments could mandate the type of interoperability remedies that in the past have protected the public from the monopoly power that first IBM and later Microsoft exercised in computer operating systems. Simply put, Facebook should be required to provide open APIs that permit third party clients to exchange status updates and share certain media such as photographs and video. The current system of email works that way, and vastly expanded the utility of the technology to users, compared to the situation where people had to have multiple email services, like bitnet, compuserve, AOL, etc., none of which connected with each other. Today people use a diverse set of email clients, and the providers of email services compete on the basis of prices, user interfaces and privacy policies. The core services people call Facebook don’t have to be a monopoly, and just like email, the sharing of status updates and media can become a more decentralized and competitive service.
Twitter can also be de-monopolized by mandating interoperable sharing of status updates, something that can allow more diversity of interfaces and policies on privacy, regulation of content, and other factors that are important to the users.
- The Commission’s remedial authority to deter unfair and deceptive conduct in privacy and data security matters;
- Evaluating the competitive effects of corporate acquisitions and mergers;
There are three areas where the FTC or DOJ evaluation of acquisitions and mergers falls short.
6.a. Free software.
The FTC and DOJ have not appreciated the role of free software in protecting the public from anticompetitive practices including but not limited to high prices and lack of interoperability.
For example, the Oracle acquisition of Sun Microsystems was opposed by several parties, on the grounds that Oracle faced competition from MySQL, a relational database program that had a very large and growing user base for Internet-based platforms. The development of MySQL was supported through a dual licensing model that included a free version, licensed under GPL, and a proprietary version. The developers of MySQL were able to charge a small number of users fees for making custom changes to MySQL, and this revenue was used to support the continued improvement of the free version, which had rapidly become a standard for many small and large web pages and web applications. KEI, the Open Rights Group and Richard M. Stallman set out objections to this merger in a letter found here: https://www.keionline.org/24887.
DOJ rejected our analysis, on the grounds that the MySQL revenues were small, compared to the primary database competitors from Oracle and Microsoft, but this argument entirely missed the point, given the massive number of MySQL users and the fact that MySQL was providing an important alternative to relational database software provided by both Oracle and Microsoft, which also disciplined prices of the Oracle and Microsoft software. As we predicted, after the acquisition Oracle effectively changed the pace and direction of the development of the free version of MySQL, so that it posed less of a threat to the more costly Oracle software. This led to greater work on other free software programs, but at significant costs in terms of delays in providing improved security, features and performance for existing installations and programs that were designed to work with MySQL.
The two lessons are (1) that importance of free software cannot be measured by its revenues, but rather by its use by and utility to users, and (2) the business model for supporting the continued development of a free software platform can be unconventional, but important and responsible for significant public benefits.
6.b. Collaborations between competitors.
A standard market concentration index such as the Herfindahl-Hirschman index (HHI) or four firm concentration index focuses on the market shares of the merging or acquired entities, in terms of services or products sold in a relevant market. In some fields, this leads to a misleading assumption that sufficient competition exists among firms post market entry. One set of facts that is often ignored in the analysis are the collaborations among firms in a market. Most important of these are joint production, transportation or marketing agreements, and licensing of intellectual property rights, including patent rights. There are many examples where such agreements are important, such as in the oil industry, where companies are often partners in the operation of oil pipelines, developing oil fields under unitisation agreements, where oil and/or gas development of a field is operated by a single company on behalf of multiple owners of individual tracts of land. Other examples where such agreements are important are among companies that cross license patents for various information technology services, software and devices, and in the area of biomedical inventions, where patents may be licensed to different parties in different geographic areas or across similar products or services.
In merger and acquisition reviews the FTC should examine and share with the public data on the extent to which firms in relevant markets have relevant joint ventures and licensing agreements that would require the companies to be partners and collaborators as well as competitors. For example, a modified HHI or four firm concentration could be an average or a weighted average of the traditional HHI or four firm indexes, and a modified one that treats collaborators as the same firm.
6.c. Pipeline products.
On February 20, 2018, the U.S. FTC approved the Celgene acquisition of Juno, without requiring divestitures, despite the fact that both firms were developing promising and potentially competing CAR T treatments for multiple myeloma. KEI’s objection to the acquisition is available here: https://www.keionline.org/26456
The Celgene acquisition of Juno allowed one company to control two competing candidates for B-cell maturation antigen (BCMA) targeted CAR T treatments for multiple myeloma. The two previous CAR T technologies approved by the FDA had initial prices of $475,000 (Kymriah) and $373,000 (Yescarta), apparently not including additional services relating to the treatment.
Both technologies that are now controlled by Celgene have benefited enormously from NIH funding. The merger significantly reduced the odds there will be robust price competition for CAR T multiple myeloma treatments, and there were zero benefits in allowing Celgene to acquire the competing BCMA CAR T treatment, given the intense investor interest in CAR T.
It is far simpler to prevent an anti-competitive acquisition than to remedy the monopolist abuses that follow. It is our understanding that the FTC is unwilling to consider pipeline products as sufficiently important to block a merger or require a divestiture. This is a mistake that the FTC needs to correct, particularly given the procedures adopted by the FTC regarding approvals of novel treatments for particularly severe illnesses.
- Evidence and analysis of monopsony power, including but not limited to, in labor markets;
- The role of intellectual property and competition policy in promoting innovation;
Patents and other intellectual property rights have very differential impacts in different fields of technology, and policies about patents should recognize these differences.
The WTO TRIPS Agreement appears to require uniformity in the patent rights across technologies, although in practice, this is probably a weaker requirement. Article 27 of the TRIPS provides that “patents shall be available . . . in all fields of technology” with “patent rights enjoyable without discrimination as to . .the field of technology. . . .” That said, there remains considerable national flexibility. Since the TRIPS Agreement came into effect in 1995, the WTO adopted the Doha Declaration on TRIPS and Public Health, which set out different requirements for the implementation of patent rights when public health is involved: “the Agreement can and should be interpreted and implemented in a manner supportive of WTO members’ right to protect public health and, in particular, to promote access to medicines for all.”[1] There is also state practice which provides for differential treatment. Governments have special rules for a variety of subject areas, including business method patents, biologic drugs, nuclear energy and seed varieties, to mention a few.
Some of the differences involve expanded rights, such as the provisions in national laws that grant patent extensions for pharmaceutical drugs or certain agricultural inventions based upon the timing of marketing approval. 35 USC § 156 requires differential treatment on patent extensions for “a patent which claims a method of manufacturing the product which primarily uses recombinant DNA technology in the manufacture of the product.”
Patent exceptions are also tailored to specific policy objectives and technologies.
The European Biotechnology Directive mandates compulsory licensing for patents on “new plant characteristics resulting from genetic engineering.”[2]
(52) Whereas, in the field of exploitation of new plant characteristics resulting from genetic engineering, guaranteed access must, on payment of a fee, be granted in the form of a compulsory licence where, in relation to the genus or species concerned, the plant variety represents significant technical progress of considerable economic interest compared to the invention claimed in the patent;
(53) Whereas, in the field of the use of new plant characteristics resulting from new plant varieties in genetic engineering, guaranteed access must, on payment of a fee, be granted in the form of a compulsory licence where the invention represents significant technical progress of considerable economic interest;
The United States has a variety of compulsory licensing statutes, including for example statutes that deal with nuclear energy, clean air, efficient lighting technologies, energy storage and biologic drugs. See: https://www.keionline.org/cl/statutory-authority-us
The notion of a single patented technology protecting a product or service is hardly relevant in some fields. The development of complex ecommerce platforms, software or mobile data and computing devices is likely to infringe large numbers of patents, which can result in significant barriers to entry, and in some cases high degrees of industry concentration.
Medical technologies
It is sometimes said that pharmaceuticals provide both the best and the worst justification for the patent system. It is certainly true that among all industries, the elimination of the patent system would have the largest impact on the pharmaceutical industry. Often the costs of manufacturing a drug or vaccine are trivial, and fixed cost of development are very large. Without a system of incentives to reward successful R&D efforts, private investments would be vastly reduced. That is one set of facts. On the other hand, drugs, vaccines and other medical technologies can be essential for health or even life. Without insulin a type 1 diabetic will die quickly, and the same is true for many other treatments for other diseases. Extending life, or reducing suffering, is important, and is an important difference for policy makers. Also, for expensive new medicines and treatments like CAR T, the patient isn’t really expected to pay for the cost of the treatment — through a patchwork and imperfect system of insurance and government programs, society is. With third parties often in charge of paying for medical technologies, and premature death and suffering a consequence of access barriers (which are significant even in the United States), the grant of a monopoly has significant costs.
The following 15 points are from a recent attempt to summarize the case for considering alternatives to the grant of monopolies on new medical technologies as the incentive to induce investments in R&D.
- People rarely stop to think of the disadvantages of linking R&D rewards for drug development to the prices of products, or consider the complexities that such approaches involve. It’s what we know, but it is a ridiculously complex and flawed system.
- Among the random elements that determine R&D rewards are relatively arbitrary patent landscapes and the various national systems of insurance coverage, which use restrictive coverage rules and co-payments to control costs.
- Under the current system, there are excessive rewards for replicating health outcomes and promoting use, even when inappropriate, and often inadequate rewards for moving science and health outcomes forward.
- Drug developers have relative high discount rates, which make it expensive to provide incentives that are earned 14 to 20 years after market entry.
- It is not uncommon for a drug to have more than one indication, including in some cases very different doses, and thus, costs to patients/payers, which have little to do with differences in outcomes. For example, midostaurin.
- Prices for treatments for rare disease are perhaps the most arbitrary, and have nothing to do with company R&D costs or the sales earned on products.
- In the area of medicine, some drugs and treatment procedures are protected by a single patent, but often there are many more asserted. For new technologies, like CAR T, or CRISPR, there are often large numbers of patents filed with overlapping and overly broad claims. Not only does the complex patent landscape for drugs create barriers for innovation, and drives up the costs of market entry and R&D, but it can result in highly arbitrary terms on monopoly, raising important questions about the relationship between the incentives provided and the objectives of creating the monopoly as an incentive in the first place.
- The sales from a new drug can range from tens of billions to tens of millions.[3] There is no effort by policy makers to consider if the distribution of returns makes any sense, given the purpose of the incentive.
- Technology assessment for determining the value of a new drug is far from an exact science, which in itself is not a fatal flaw, until it is linked to decisions that determine access to a life saving treatment.
- The costs of the current system in terms of under serving populations that would benefit from treatments is rarely measured. How many women are dying because they do not have access to TDM1/Kadcyla for example?
- If and when governments delink R&D incentives from prices, they can provide far more rational reward systems, using existing data on outcomes and budget constraints, and vastly expand access and reduce inequalities.
- Under delinkage systems, like the one proposed in S.495, rewards can be targeted to induce investments in treatments that improve outcomes (Sec 9) , create priorities (Sec 10), and advance science (Sec 11, Sec 12).
- The often heard argument that the current system “works” are shallow attempts to excuse flaws and avoid even thinking about alternatives that would work better.
- It’s not because a plumber can fix a leaking pipe the cost is reasonable. It’s not because a mechanic can fix a car the bill is reasonable. It’s not because a day in a hospital saved a life the cost is reasonable. Why should new drugs be different?
- Aside from the harsh impacts of fiscal toxicity for patients who receive treatments, there is considerable inequality of access, based upon incomes and geography. This inequality is the opposite of evidence the current system “works.” When there is no real plan to address inequalities of access, there is evidence policy makers are not serving the underserved, and instead are protecting those that are most privileged, and benefit the most from the current system.
Policy makers need to explore both short term and longer term reforms, as regards the incentives for development of new drugs and other medical technologies, like CAR T.
In the near to medium term, policy makers should reform the system of exclusive rights so that incentive is more rationally related to policy objectives.
Non-patent exclusivities have fixed terms that are consistent, but often the costs are spectacularly random when compared to the benefits, such as the very wasteful pediatric testing exception, which can cost more than $5 million per child tested, or the orphan drug exclusivity, which is sometimes used to drive up the cost of existing drugs for new indications where the costs of testing was minimal, and for new drugs that generate billions of revenue per year.
Patents may have uses in rewarding medical inventions, but they are a rough instrument for shaping incentives for drug development. The science for drug development is often moved forward by government-funded research, and the more important role for the private sector is to provide investments in costly clinical trials and regulatory approval, areas where patents usually play a minor role. In the current system, you have cases where a drug that is costly to develop has no patents, and cases where a drug that is relatively cheap, with few patients in trials, has a large number of patents. Some products obtain patents of each new use, formulation and use in a combination, and can put off competition for many years, based simply on the skills and ingenuity of patent lawyers and the often arbitrary decisions by juries asked to revolve highly technical patentability issues.
For biologic drugs the patent landscape is actually treated as a protected secret by the misnamed “Biologics Price Competition and Innovation Act of 2009 (BPCIA),” a ridiculous policy that nullifies one of the putative benefits of the patent system — technology transfer. And, technology transfer is badly needed for biologic drugs.
KEI recently studied competition for drugs registered with the FDA from 1995 to 2005. It found that 17 percent of new BLA drugs and 61 percent of new NDA drugs faced competition from at least one product with the same API by the end of 2017. A small molecule drug was 3.5 times more likely to face any competition than a biologic drug, but that’s not all. The first entrant for a small molecule occured an average of 6 years later than was the case for a biologic. The number of companies selling a drug with the same API was also quite different. There were an average of 9 companies approved to sell drugs with the same API when the drug was a small molecule. For biologic drugs, the average number of companies selling a drug with the same API was 1.5, for the drugs that faced any competition at all. On top of everything else, physicians are less willing to prescribe biosimilar drugs, over concerns they may not work the same.
Clearly, the whole system works very differently for small molecules than for biologic drugs, leading to fewer competitors for biologic drugs, longer terms of monopolies, and less price competition, even though the R&D costs are similar.
To fix this lack of competition for biologic drugs, policy makers need to force technology transfer, which would not only enhance competition, it would assure patients the biosimilar drugs would work, and present less risk to patients.
We propose the following obligations:
As a condition of registration of biological products and services a person must agree to promptly, upon request, make available to providers of generic or biosimilar products or services certain materials, data, information and know-how, relating to the manufacture or supply of the regulated product, including but not limited to, when appropriate and relevant, the following:
- a. Materials:
- i. Cellular clones and hybridoma stocks;
- ii. Plasmids, plasmid maps, and sequences of antibody complementarity determining regions (CDR); and
- iii. Physicochemical/biophysical characterization;
- b. Methods:
- i. Growth conditions and protocols;
- ii. Attenuation or inactivation protocols;
- iii. Extraction and purification protocols; and
- iv. Synthetic work-up and schemes;
- c. Sufficient quantities of the approved medication for testing, and the protocols/methods used for testing the products, and the expected outcomes from those protocols.
Reforms of the incentive system should seek a closer match between the incentive and the costs of investments the incentive is designed to stimulate or reward. One approach proposed to the NIH, for government-owned inventions, is to reduce the period of exclusivity when a product meets certain global revenue benchmarks.
The notion that the period of exclusivity should be almost random depending upon the patent landscape, and that the amount of revenue earned under the monopoly also differs radically without regard to the expected risk-adjusted investments in R&D, should not be unquestioned or unexamined. Modeling approaches such as those proposed by KEI for several NIH proposed exclusive licenses noticed in 2018 (see: https://www.keionline.org/nih-licenses) would be useful, to see if reducing exclusivity or drug prices after products exceed certain revenue benchmarks would make the incentives more efficient and cost effective.
More important, however, is to model approaches that delink the R&D incentives from the prices of products or services. The National Academies is keen to undertake a feasibility study of delinkage, including the transition from the current system, so that a progressive delinkage of R&D incentives from prices has a feasible and cost effective path.
The federal government can do much to improve the transparency of R&D costs by publishing the costs of clinical trials on products subsidized by or licensed from the federal government. It is astonishing that the NIH and other federal agencies such as BARDA refuse requests to provide information on the costs of clinical trials subsidized by the federal government.
The NIH and the Army have both refused to require that companies licensing patents that have federal Bayh-Dole rights refrain from charging prices in the United States that are higher than the companies charge in other high income countries, thus endorsing a policy of discriminating against the United States, when products are subsidized by U.S. taxpayers. This harms U.S. taxpayers, employers and patients.
- The consumer welfare implications associated with the use of algorithmic decision tools, artificial intelligence, and predictive analytics;
- The interpretation and harmonization of state and federal statutes and regulations that prohibit unfair and deceptive acts and practices; and
- The agency’s investigation, enforcement and remedial processes.
In 2014, KEI asked the FTC to investigate collusion between Sanofi and Shire, relating to the 2012 decision by the Icahn School of Medicine at Mount Sinai (ISMMS) to license Fabry patents to Shire in Europe, and the subsequent decision by Shire to withdraw an FDA biologic license application (BLA) for Replagal (agalsidase alfa). Our letter setting out the basis for the complaint is here: https://www.keionline.org/22538. KEI believes there was strong evidence of collusion to limit competition between Sanofi, which sells Fabrazyme, and Shire, whose treatment Replagal is Fabryzyme’s direct competitor. To our knowledge, the FTC’s investigation was brief and shallow, and did not involve discovery. There is simply no other reason than collusion to understand why Shire’s Replagal has not entered in the U.S. market for the treatment for Fabry’s disease, a serious illness with an enormous price tag for the treatments. Fabrazyme and Replagal were both invented on NIH grants, and both products are now owned by European firms. The U.S. prices for Fabrazyme are extremely high, and would be lower if Sanofi and Shire were acting as competitors, rather than companies managing a global cartel.
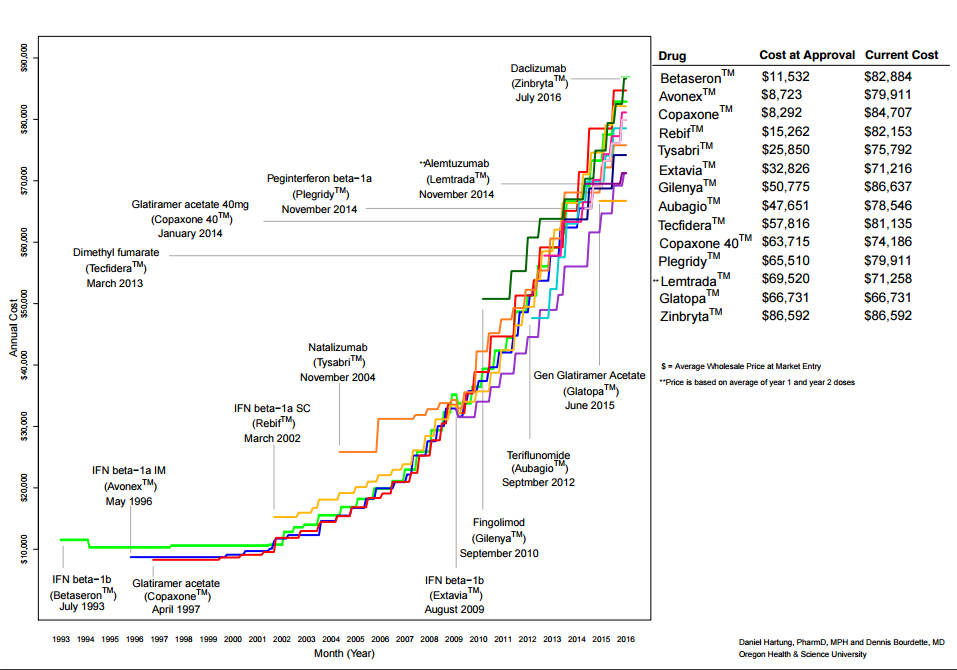
We are anxious to see investigations of potential pricing collusion in the markets for insulin and drugs for multiple sclerosis (MS), two areas where prices and price increases have clearly moved in concert.
The attached figure prepared by Daniel Hartung and Dennis Bourdette at Oregon Health and Science University illustrates how a series of drugs to treat MS have seen price escalations[4] to match the ever-increasing rollout prices of new drugs. R&D costs are clearly not a factor. In the figure, the highest price for a drug and the one with the largest increase for a rollout price was Zinbryta, a repurposed drug licensed from the NIH to Biogen for modest consideration. Zinbryta was later withdrawn from the market over concerns regarding its safety,[5] but the fact that the NIH allowed its own patent to be licensed for a product sold at such a high price illustrates the extent that the federal government has failed to exercise its own leverage to curb excessive prices. The FTC could fruitfully engage in advocacy, not only to influence the terms of licenses on government-owned patents, but to set norms for the use of Bayh-Dole march-in or royalty-free rights in drugs invented on federal grants or research contracts.
Figure 1: Price increases for drugs treating multiple sclerosis
Endnotes:
[1] Paragraph 4. Doha WTO Ministerial 2001: TRIPS, WT/MIN(01)/DEC/2, 20 November 2001. https://www.wto.org/english/thewto_e/minist_e/min01_e/mindecl_trips_e.htm
[2] Directive 98/44/EC of the European Parliament and of the Council of 6 July 1998 on the Legal Protection of biotechnological inventions.
[3] See: http://drugdatabase.info/revenues/
[4] Hartung, Daniel M. et al. “The Cost of Multiple Sclerosis Drugs in the US and the Pharmaceutical Industry: Too Big to Fail?” Neurology 84.21 (2015): 2185–2192. PMC. Web. 20 Aug. 2018. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4451044/pdf/NEUROLOGY2014614974.pdf